 |
|
|
Protein-water interfaces |
A central goal of molecular biology is to understand how cells work. This, of course, requires a detailed
understanding of the cell`s constituting parts, such as proteins. These biological macromolecules, which fulfill a huge
variety of different functions in the cell, have been likened to molecular machines. To unravel how the function, one obvious
prerequisite is knowledge of their structure. Yet, purely structural information is probably not enough. Since proteins are
flexible, studying the kinetics and energetics of reaction intermediates is important.All interactions, in particular those
between ligands and proteins, are mediated by solvent. This is especially relevant for electrostatic forces, whose central
role in biological processes is well established. The roles and properties of water in a protein solution vary significantly,
but can be rougly categorized as follows:
- Individual water molecules can be tighly bound and even buried within the protein. Frequently, these bound waters fulfill
an improtant functional role.
- There are bulk waters which behave (almost) as molecules in the neat liquid.
- Finally, the solute is surrounded by and strongly interacts with so-called ordered or hydration water molecules.
Considering the difficulties of purely experimental approaches, molecular dynamics simulations can help interpret experimental
results, such as neutron diffraction, dielectric relaxation, and NMR cross-relaxation experiments. Further simulations yield
additional information, such as radial distribution functions and orientational correlation between the protein and the
solvent. Dipole-dipole and ion-dipole correlation functions give a deep insight in the interactions of proteins and their
solvents.
 Furthermore, these properties can be examined for bulk and hydration
water seperately by dividing the solvent molecules into solvent shells around the protein. Two solvent shells could be clearly
discerned about charged and polar amino acids. Furthermore, these properties can be examined for bulk and hydration
water seperately by dividing the solvent molecules into solvent shells around the protein. Two solvent shells could be clearly
discerned about charged and polar amino acids.
We studied the mean residence time of water molecules for water shells about the full protein, as well as for water layers
about individual amino acids. In the dynamic properties, two solvent shells could be characterized as well.
However, by comparison to simulations of pure water it could be shown that the influence of the protein reaches beyond 6 Å,
i.e., beyond the first two shells. In the first shell (r < 3.5 Å), the structural and dynamical properties of solvent
waters varied considerably and depended primarily on the physico-chemical properties of the closest amino acid side chain,
with which the waters interact.
By contrast, the solvent properties seem not to depend on the specifics of the protein studied (such as the net charge) or
on the secondary structure element in which an amino acid is located. While differing considerably from the neat liquid,
the properties of waters in the second solvation shell (3.5 Å < r <6 Å) are rather uniform;
direct influence from surface amino acids is already mostly shielded.
The calculation of the frequency dependent dielectric constant of a solution is based on the time correlation function of the
net dipole moment of the system. The dielectric susceptibility can be directly compared to experimentally measured frequency
dependent dielectric constants. Our approach, to which we refer as dielectric component analysis, starts from the
observation that the net dipole moment of the system can be decomposed into individual contributions. One straightforward
possibility is to write the dipole moment as the sum of the dipole moment of the protein plus the dipole moment of all water
molecules in all solvent shells. Because of the linearity of the Fourier-Laplace transform, each term gives rise to a
component susceptibility. The interpretation of dielectric properties just discussed is based on an analysis of regional
electric dipole moments. Thus, the study of dielectric relaxation operates on a mesoscopic scale. By contrast, NMR based
relaxation techniques operate on the molecular scale since the method, in principle, detects and traces the magnetic dipole
moments of individual nuclei. The central quantity of NMR experiments exploiting the Nuclear Overhauser effect is the
correlation function:
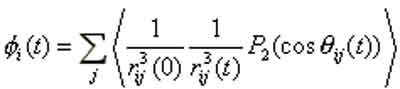 The locality of the Nuclear Overhauser correlation function makes it extremly useful for NMR based structure determination of
biomolecules.
The locality of the Nuclear Overhauser correlation function makes it extremly useful for NMR based structure determination of
biomolecules.
|
 To the top To the top |
|

