Intramolecular vibrational energy redistribution (IVR) is an important step of reactive processes in chemistry. The
assumption of rapid IVR inside a molecule prior to reaction, on the one hand, is essential for the applicability of
statistical rate theories. If randomisation of vibrational energy is hindered by internal bottlenecks such that reaction is
faster than IVR, on the other hand, the system behaves nonergodically which allows for mode specific chemistry. Hence, great
efforts have been expended over the last decade to elucidate the mechanisms and principles determining IVR.
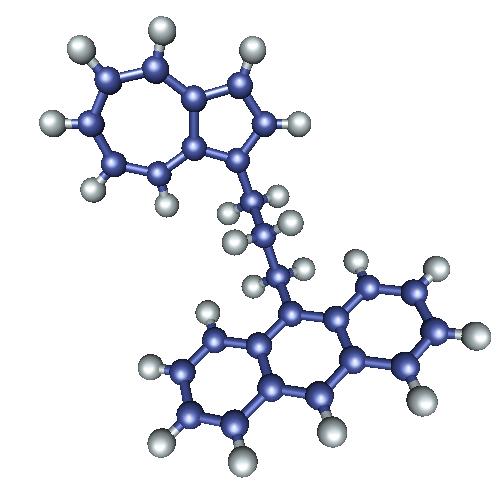 In our group we focused on the vibrational energy transfer through
molecular chains which at each side are connected to relatively large chromophores which also act as heat reservoirs. In the
experiment the azulene part is vibrationally excited by a short laser pulse; subsequently the time for reaching intramolecular
equilibrium, by transfer through the molecular chin to the other chromophore, is measured. Our measurements of vibrational
energy transfer through molecular chains in principle resembles a classical macroscopic heat conduction experiment in which
two heat resvervoirs of different temperature are connected by a heat conducting rod. The influence of anharmonicity , disorder,
system size and presence of external potentials on the energy transport properties of these chains has been investigated by
molecular dynamics. In our group we focused on the vibrational energy transfer through
molecular chains which at each side are connected to relatively large chromophores which also act as heat reservoirs. In the
experiment the azulene part is vibrationally excited by a short laser pulse; subsequently the time for reaching intramolecular
equilibrium, by transfer through the molecular chin to the other chromophore, is measured. Our measurements of vibrational
energy transfer through molecular chains in principle resembles a classical macroscopic heat conduction experiment in which
two heat resvervoirs of different temperature are connected by a heat conducting rod. The influence of anharmonicity , disorder,
system size and presence of external potentials on the energy transport properties of these chains has been investigated by
molecular dynamics.
The intramolecular energy flow does not depend on whether the claculations are done for the isolated molecule or for the
azulene-anthracene compound dissolved in a bath of Xe. Therefore, most of the calculations were done for the isolated molecule.
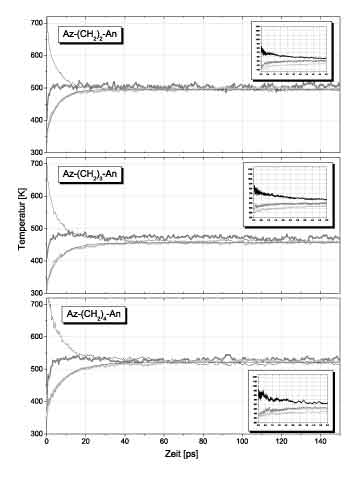 In the left figure a typical result of a MD simulation is shown. The black curve belongs to the azulene part, the dark grey
one to the chain and the light curve to anthracene. From the kinetic vibrational energies of the atoms the temperatures
of different parts of the molecule were calculated and plotted versus time. In accord with the expermental observation
intramolecular equilibration proceeds in two steps. There is a fast subpicosecond component caused by harmonic energy flow
followed by the IVR process taking several picoseconds. The harmonic energy flow is most clearly seen for the azulene
temperature which within the first 300 fs decays from approximately 800 K to 700 K. This corresponds to a loss of 20% of the
excitation energy and is in complete agreement with a harmonic mode simulation. Subsequently intramolecular energy
equilibration takes places leading to an equlibrium temperature of 450 K. IVR can be described by single exponential curves.
The calculated time constants were about 50% larger than the experimental values. Most interestingly, however, the dependece
of the IVR - timeconstant on the length of the bridge showed the same trends as found in the experiment. In particular, for n>4
CH2-units the time for IVR became constant and independent of the chain length such as demonstrated in Fiure 2.
In the left figure a typical result of a MD simulation is shown. The black curve belongs to the azulene part, the dark grey
one to the chain and the light curve to anthracene. From the kinetic vibrational energies of the atoms the temperatures
of different parts of the molecule were calculated and plotted versus time. In accord with the expermental observation
intramolecular equilibration proceeds in two steps. There is a fast subpicosecond component caused by harmonic energy flow
followed by the IVR process taking several picoseconds. The harmonic energy flow is most clearly seen for the azulene
temperature which within the first 300 fs decays from approximately 800 K to 700 K. This corresponds to a loss of 20% of the
excitation energy and is in complete agreement with a harmonic mode simulation. Subsequently intramolecular energy
equilibration takes places leading to an equlibrium temperature of 450 K. IVR can be described by single exponential curves.
The calculated time constants were about 50% larger than the experimental values. Most interestingly, however, the dependece
of the IVR - timeconstant on the length of the bridge showed the same trends as found in the experiment. In particular, for n>4
CH2-units the time for IVR became constant and independent of the chain length such as demonstrated in Fiure 2.
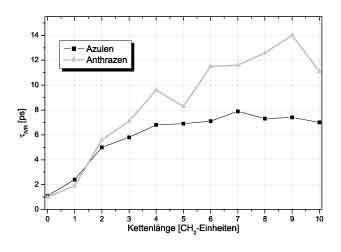 Figure 2: IVR relaxation times as function of chain length Figure 2: IVR relaxation times as function of chain length
For molecules with shorter chains, we observe strong temperature gradients not only at the azulene but also at the athracene
side, indicating that the coupling of the chromophores to the bridge is limiting the energy flux. In the chain the energy
transport obviously proceeds much faster and, therefore, the time needed to distribute the vibrational energy over the whole
molecule with increasing chain length remains the same for n>4. This can be consistently be explained by assuming ballistic
energy transport in the chain connectiong both chromophores. The strong temperature gradient in Figure 3 extends over
2-3 bonds which might be the reason, that, for shorter bridges, the IVR time constant is proportional to the chain length.
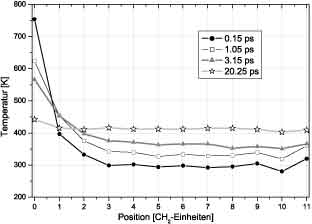 Figure 3:Temperature gradient in the aliphatic chain Figure 3:Temperature gradient in the aliphatic chain
Literature :
submitted to Journal of Chemical Physics
|

