 |
|
|
Energy transfer of CH2I2 |
Intramolecular vibrational redistribution (IVR) and intermolecular vibrational energy transfer (VER) are undoubtedly
of fundamental importance in chemical reaction dynamics. The validity of statistical reaction rate theories, such as the
microcanonical RRKM or canonical transition state theories, depends crucially on how rapidly an equilibrium is established
between the nonreactive modes or solvent and the chemical reaction coordinate. Therefore, IVR has been investigated under
isolated molecule conditions and in low pressure gases for quite some time and can be considered a relatively mature field of
research in chemical physics, both with respect to experiment and theory.
In studies of large polyatomics, owing to the large density of states, it is almost always assumed that the vibrational
excess energy is nearly statistically distributed among all modes on the timescale of VET (rapid IVR). Small polyatomic
molecules, on the other hand, due to a substantially smaller density of states, at least for low to moderate excitations,
offer the possibility to slectively excite and interrogate certain vibrational motions and therefore to gain mode specific
information on vibrational energy flow in molecules and solutions. Moreover, due to slower IVR, one can hope to dissentangle
intramolecular and intermolecular energy relaxation processes.
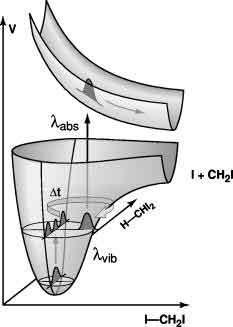 Diiodomethane CH2I2 was examined in CCl4 and CDCl3 solutions at 300 K by molecular
dynamics simulations. A combination mode of the
symmetric and asymmetric C-H-stretch mode of the solute was excited. The force field was designed to mimic the experimental
vibrational frequencies of CH2I2 in the gas phase. The overall energy relaxation to the solvent is
exponential to a very good approximation, the associated relaxation time being 23 ps in CDCl3 and 117 ps in
CCl4 respectively. Turning to normal mode vibrational energies, one observe that energy transfer from the
initially excited modes to lower frequenc dominantly C-H-vibrations is very rapid ( with times of the order of 1 ps or
below ). For the lower frequency vibrations mostly localized on the CI2 moiety, including the Franck-Condon active
mode, energy rise times are longer by factors of 2 to 20 as compared to energy population times observed for the higher
frequency C-H-modes. Interestingly, the vibrational energy of the lowest frequency vibration ( 122 cm-1,
CI2 bending ) stays close to equilibrium for all times. Seemingly, this mode is merely a spectator in the overall
process of vibrational redistribution and relaxation, thereby questioning the general concept of lowest frequency doorway
vibrations.
Diiodomethane CH2I2 was examined in CCl4 and CDCl3 solutions at 300 K by molecular
dynamics simulations. A combination mode of the
symmetric and asymmetric C-H-stretch mode of the solute was excited. The force field was designed to mimic the experimental
vibrational frequencies of CH2I2 in the gas phase. The overall energy relaxation to the solvent is
exponential to a very good approximation, the associated relaxation time being 23 ps in CDCl3 and 117 ps in
CCl4 respectively. Turning to normal mode vibrational energies, one observe that energy transfer from the
initially excited modes to lower frequenc dominantly C-H-vibrations is very rapid ( with times of the order of 1 ps or
below ). For the lower frequency vibrations mostly localized on the CI2 moiety, including the Franck-Condon active
mode, energy rise times are longer by factors of 2 to 20 as compared to energy population times observed for the higher
frequency C-H-modes. Interestingly, the vibrational energy of the lowest frequency vibration ( 122 cm-1,
CI2 bending ) stays close to equilibrium for all times. Seemingly, this mode is merely a spectator in the overall
process of vibrational redistribution and relaxation, thereby questioning the general concept of lowest frequency doorway
vibrations.
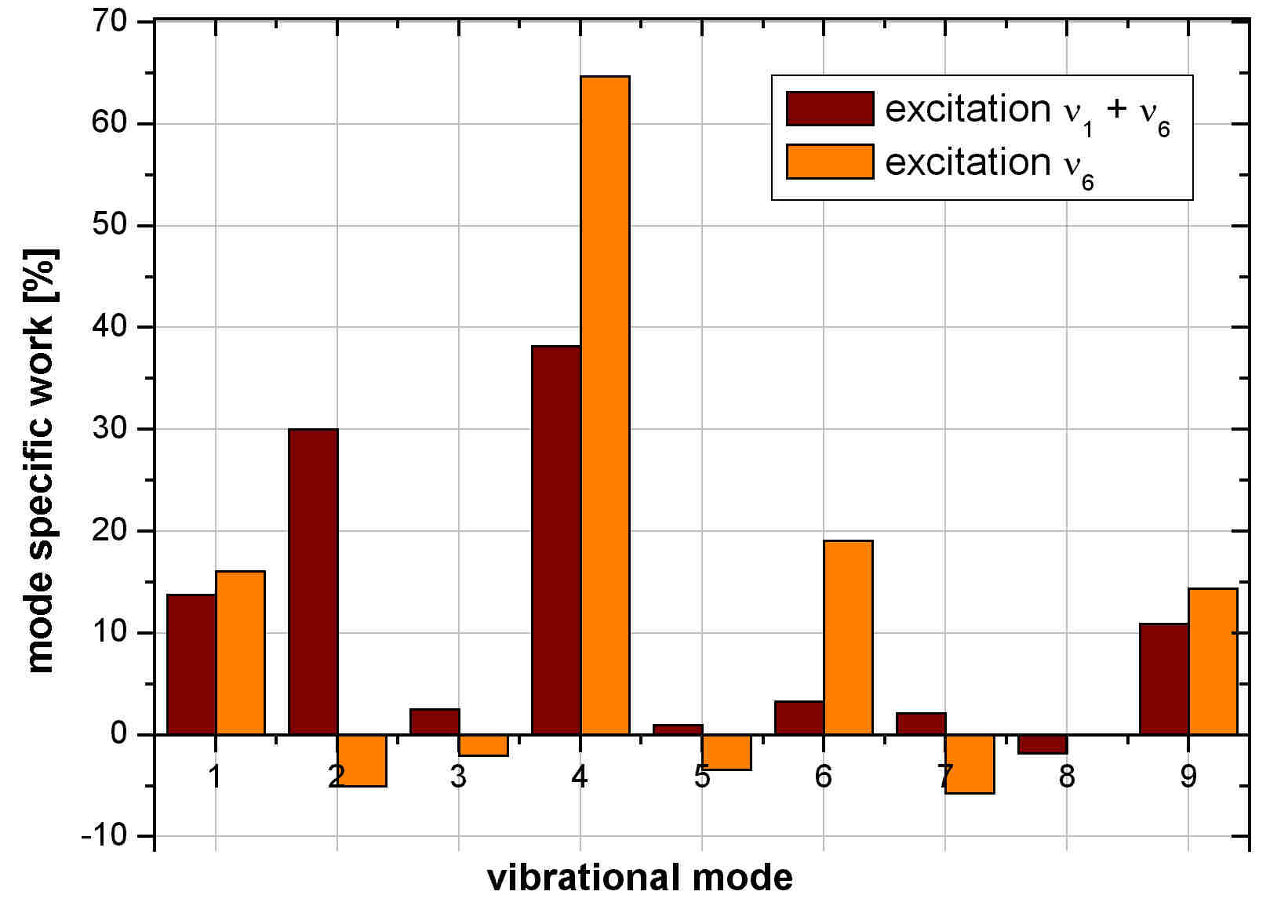 The figure above contains the mode specific intermolecular work done by the environmental degrees of freedom
upon the vibrational normal modes. These work terms correspond to the direct solute-solvent energy flow via the respective
vibrations. The largest part of the vibrational excess energy is transferred to the solvent via the C-H-rocking mode (see figure
below).
The figure above contains the mode specific intermolecular work done by the environmental degrees of freedom
upon the vibrational normal modes. These work terms correspond to the direct solute-solvent energy flow via the respective
vibrations. The largest part of the vibrational excess energy is transferred to the solvent via the C-H-rocking mode (see figure
below). Intermediate vibrations make less contributions. These work terms reflect effects of restricted IVR, due to slow population
of the lowest frequency vibration (IVR bottleneck), as well as the different effenciencies of normal mode vibrations in the
intermolecular vibrational relaxation at restricted microcanonical equilibrium.
Intermediate vibrations make less contributions. These work terms reflect effects of restricted IVR, due to slow population
of the lowest frequency vibration (IVR bottleneck), as well as the different effenciencies of normal mode vibrations in the
intermolecular vibrational relaxation at restricted microcanonical equilibrium.
Literature :
Phys. Chem. Chem. Phys., 4, 271-278 (2002)
|
 To the top To the top |
|

